
Gå tillbaka
Mekanismer för reglering av insulinfrisättning
KATP-BEROENDE OCH -OBEROENDE MEKANISMER FÖR REGLERING AV GLUKOS-STIMULERAD INSULINFRISÄTTNING
Implikationer
för behandling av sviktande insulinfrisättning vid typ 2 diabetes
av Hindrik Mulder, Leg.läk.,
Institutionen för cell-och molekylärbiologi Avdelningen för
molekylär signalering, Lunds universitet, BMC C11, 221 84 Lund
046-2224022,
hindrik.mulder@medkem.lu.se
Det har länge rått en närmast total samstämmighet vad gäller den avgörande rollen metabolism av glukos i ß-cellen spelar för insulinfrisättning. En s.k. konsensus-modell har formulerats, vari de flesta komponenter har identifierats och karakteriserats. Emellertid har man nyligen beskrivit en kompletterande mekanism för hur glukos reglerar frisättning av hormonet. Eftersom denna mekanism till viss del utnyttjar en annan signalväg, som endast delvis är känd, kan det framöver öppna sig nya möjligheter för att terapeutiskt angripa den sviktande insulinsekretionen vid typ 2 diabetes. I det följande ges en översikt av dessa nya framsteg och hur de förhåller sig till den vedertagna konsensus-modellen.
Konsensus-modellen
den KATP-beroende vägen för glukos-stimulerad insulinfrisättning.
Processen som kontrollerar frisättning av insulin kan för enkelhetens skull delas in i en metabol fas, en elektrisk fas och en exocytotisk fas. De två första faserna är till stor del unika för insulincellen medan exocytos är ett mera generellt fenomen som förkommer i många olika celltyper. Ur ett funktionellt perspektiv är det just egenskapen att metabolismen i insulincellen kontrollerar exocytos av insulin som förläner cellen dess speciella betydelse. De molekylära skeendena i konsensus-modellen illustreras i figur 1.
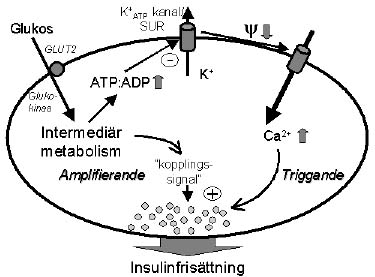
Figur 1. Konsensus-modellen beskriver den KATP-beroende vägen för glukos-stimulerad insulinfrisättning. I schemat har också en förmodad "kopplingssignal" adderats; denna emanerar från glukosmetabolismen och ansvarar för den KATP-oberoende vägen för glukos-stimulerad insulinfrisättning och griper in någonstans distalt om KATP-kanalen. KATP-kanal ATP-känslig K+ kanal; SUR sulfonylureareceptor; GLUT2 glukostransportör 2; Y - membranpotential
ß-cell metabolism och insulinfrisättning
Den metabola fasen inleds av att insulincellen (eller ß-cellen) känner av förändringar i blodglukos genom en koordinerad insats av två proteiner glukostransportör 2 (GLUT2) och glukokinas. Detta glukosensorbegrepp har ersatt äldre, och inkorrekta, föreställningar om en glukoreceptor i ß-cellmembranet. GLUT2 förmedlar en faciliterad diffusion av glukos över membranet in i ß-cellen varvid sockret fosforyleras av glukokinas.
Detta enzym är i själva verket ett av enzymerna i hexokinasfamiljen och benämns ibland hexokinas IV. Glukokinas har speciella egenskaper som gör att det närmast är skräddarsytt för sin roll som glukosensor och hastighetsreglerande steg i glukos-metabolismen i ß-cellen. Till skillnad från de övriga hexokinaserna är glukokinas inte föremål för en allosterisk hämning av glukos-6-fosfat. Om så vore fallet skulle glukosmetabolismen stanna upp vid höga glukosnivåer, vilket inte är önskvart av en mekanism som skall reglera sekretion av ett hypoglykemiskt hormon. Vidare har glukokinas ett Km för glukos som ligger nära den fysiologiska koncentrationen av sockret i blodet. Detta innebär att enzymet kommer att kunna förändra sin aktivitet som svar på fysiologiska blodsockerfluktuationer. Övriga hexokinaser har ett Km som ligger i storleksordningen av µM, vilket betyder att de är maximalt aktiva vid subnormala glukos-koncentrationer och därmed oförmögna att känna av fysiologiska förändringar i blodsocker. Betydelsen av de inledande stegen i glukosmetabolismen och glukokinas för insulinfrisättning understryks av att vi idag har detaljerad kunskap om hur insulinbrist uppstår vid MODY maturity onset diabetes of the young. Denna diabetesform omfattar 5-10% av alla patienter med typ 2 diabetes och debuterar vid yngre åldrar än den idiopatiska typen. En subgrupp av dessa patienter bär på en muterad allel i glukokinasgenen, vilket resulterar i en förlångsammad glykolys i b-celler. Härvidlag hämmas den metabola signalering som kopplar stimulus (glukos) till sekretion (insulin), b-cellerna frisätter ej adekvata mängder insulin som svar på förändringar i blodglukos och hyperglykemi utvecklas. Sammantaget är ett resultat av metabolismen av glukos i glykolys och citronsyra-cykel att cellens energiladdning ökar. Detta kan uttryckas som att den cellulära ATP: ADP kvoten stiger och anses vara den kritiska metabola signalen som initierar eller triggar insulinfrisättning.
ß-cellen en elektriskt excitabel cell
I b-cellen upprätthålls en negativ membranpotential till stor del av en K+-kanal som låter jonen strömma ut ur cellen längs den elektrokemiska gradienten. Kanalen stängs av ATP och hålls öppen av Mg2+-ADP; den benämns KATP-kanalen.
KATP-kanalens egenskaper tillåter metabolismen i b-cellen, via produktion av ATP (ATP:ADP kvot stiger), att kontrollera membranpotentialen. Således, när kanalen stängs på grund av inbindning av ATP minskar K+-strömmen över membranet som därvidlag depolariseras.
Härmed öppnas en spännings-känslig Ca2+-kanal, Ca2+rusar in i cellen från i huvudsak extracellulärrummet och den elektriska fasen av insulinfrisättningen avslutas. KATP-kanalen byggs upp av en kanaldel, kir 6.2 (K+-inward rectifying), och en regulatorisk del, den så kallade sulfonylureareceptorn (SUR). Detta är viktigt att känna till eftersom de enda farmaka i bruk som direkt stimulerar insulinfrisättning är sulfonyl-urea-preparaten. De verkar genom att binda till SUR, vilket leder till stängning av KATP-kanalen och insulinfrisättning enligt de mekanismer som har beskrivits ovan.
Detta förhållande innebär också att sulfonylurea-preparaten inte är beroende av glukos för sin effekt. En negativ konsekvens av detta är att preparaten kan ge upphov till hypoglykemier eftersom insulin kan frisättas även om blodsocker skulle vara lågt, till exempel om patienten har hoppat över en måltid.
Exocytos från ß-cellen
Den exocytotiska fasen av insulinfrisättning triggas av Ca2+ inflödet i cellen. Trots att detta är väletablerat uteblir Ca2+ stegringen ses ingen exocytos och även kan observeras i samband med frisättning av hormoner och transmittorer från andra celler vet man förhållandevis lite om hur Ca2+ stegringen översätts till exocytos.
I första hand har man diskuterat betydelsen av proteinkinaser i detta sammanhang; de anses kunna aktiveras av Ca2+ stegringen och härefter modifiera målproteiner i exocytosmaskineriet. Det har emellertid inte varit enkelt att identifiera dessa målproteiner. Vidare är det inte heller säkerställt att de aktuella proteinkinaserna verkligen aktiveras när b-celler exponeras för glukos. I första hand är det proteinkinas A, proteinkinas C och kalcium-kalmodulinkinas II som har undersökts. Proteinkinas A aktiveras vanligen av cAMP; huruvida cAMP stiger i b-celler efter stimulering med glukos är kontroversiellt. Däremot synes proteinkinas A spela en avgörande roll för hur inkretinhormonet glukagonlik-peptid 1 (GLP-1) potentierar glukos-stimulerad insulinfrisättning.
Exocytos av insulin-innehållande granula är en komplex process som innefattar förflyttning av granula från olika intracellulära pooler, modifiering av proteiner i såväl granula- som cellmembranet, förändring av miljön inne i granula och återanvändning av membran från frisatta granula. Exempelvis vet vi idag att endast en liten fraktion av insulingranula är redo eller primade för frisättning i varje givet ögonblick; efter stimulering av b-cellen rekryteras granula från en reservpool. Att protein-proteininteraktioner är kritiska i dessa sammanhang är uppenbart och flera av de viktigaste aktörerna har identifierats.
Den KATP-oberoende vägen för glukos-stimulerad insulinfrisättning
Även om konsensus-modellen för glukos-stimulerad insulinfrisättning täcker in de flesta aspekter av hur insulinfrisättning regleras finns frågetecken. Först och främst har det länge varit känt att b-cellen desensitiseras för Ca2+. I klartext betyder detta att Ca2+ ensamt inte kan upprätthålla insulinfrisättning när b-cellen stimuleras med glukos, utan ytterligare faktorer krävs. Vidare har man också länge vetat att glukos och sulfonylurea har en additiv effekt på insulinfrisättning.
I denna situation är KATP-kanalen stängd och mekanismerna som beskrivs av konsensus-modellen maximalt utnyttjande. Trots detta kan glukos i närvaro av sulfonylurea åstadkomma ytterligare frisättning av insulin, en mekanism som skulle kunna betecknas KATP-oberoende. Den KATP-oberoende vägen för glukos-stimulerad insulinfri sättning har sedermera kunnat studeras i mer detalj tack vare en serie banbrytande försök som gjorts parallellt i Jean-Claude Henquins och Toru Aizawas laboratorier.
Grunden för dessa försök är att membrandepolarisation och därmed Ca2+ inflöde åstadkoms genom att ß-cellen depolariseras med en ökning av K+ koncentrationen i försöksbufferten. Härvidlag minskar den elektrokemiska gradienten för jonen över membranet som inte kan hållas polariserat. De spännings-känsliga Ca2+-kanalerna öppnas och en Ca2+ stegring åstadkoms utan att KATP-kanalen har varit involverad. Ofta används diazoxid en insulinfrisättningsblockerande drog som håller KATP-kanalen öppen och därmed membranpotentialen negativ för att säkerställa att inga effekter som observeras ändå åstadkoms genom förändringar av KATP-kanalens aktivitet. Det är betydelsefullt i sammanhanget att understryka att den KATP-beroende och -oberoende vägen för glukos-stimulerad insulinfrisättning opererar samtidigt; visserligen krävs en hög, ofysiologisk extracellulär K+ koncentration under experimentella betingelser för att demaskera den KATP-oberoende vägen, betingelser som varken behövs eller finns under normala förhållanden.
I de inledande studierna fann man att en hög extracellulär K+ koncentration resulterar i en ökad basal frisättning av insulin, vilket är förenligt med en depolarisation av cellmembranet. Emellertid, ökas glukos i bufferten ses ändå en förstärkning av insulinfrisättningen; denna förstärkning sker oavsett om diazoxid är närvarande eller ej, vilket betyder att glukos förmår frisätta insulin oberoende av KATP-kanalen. Dessa fenomen illustreras i figur 3 av egna försök gjorda i en insulin-producerande cellinje med hög glukoskänslighet. Den KATP-oberoende vägen för glukos-stimulerad insulin-frisättning har också beskrivits i humana öar.
(se fig. 3)
Metabola kopplingssignaler vid KATP-oberoende vägen Upptäckten av den KATP-oberoende vägen för glukos-stimulerad insulinfrisättning har varit startskottet för en intensiv jakt på vilken eller vilka metabola signaler i ß-cellen som kopplar stimulus till sekretion.
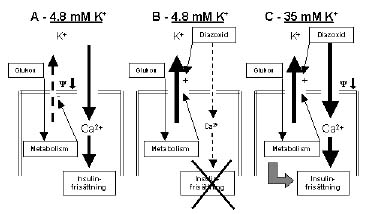
Figur 2. Experimentella betingelser varunder man kan studera den KATP-oberoende vägen för glukos-stimulerad insulinfrisättning. A Vid normalt extracellulärt K+ stänger glukos-metabolismen KATP-kanalen varvid membranet depolariseras och exocytos av insulin sker. B Diazoxid blockerar denna process genom att förhindra stängning av KATP-kanalen. C Om extracellulärt K+ höjes depolariseras membranet oberoende av KATP-kanalen. Under dessa betingelser reglerar glukos fortfarande insulinfrisättning. Detta hämmas ej av närvaro av diazoxid och är alltså KATP-oberoende. Y - membranpotential
En förutsättning är att glukos metaboliseras i ß-cellen. Huruvida Ca2+ krävs är ej helt klarlagt; en studie visar att glukos kan stimulera insulinfrisättning helt i frånvaro av Ca2+ om proteinkinas A och C samtidigt är aktiverade. En het kandidat för signalering i den KATP-oberoende vägen för glukos-stimulerad insulinfrisättning har länge varit acyl-CoA, den intracellulära, aktiverade formen av fettsyror.
När glukos metaboliseras i ß-celler ställer cellen om från oxidering av fett till syntes av lipider (esterifiering); denna omställning åstadkoms genom att förhöjda nivåer av malonyl-CoA uppkommer som ett resultat av glukosmetabolism. Malonyl-CoA stänger av oxidation samtidigt som den själv utgör substrat för fettsyrasyntes. Härmed ökar poolen av acyl-CoA i cellen och dessa lipider har föreslagits att fungera som metabola signaler.
En studie visar att fettsyror härmar effekten av glukos vid KATP-oberoende betingelser. I våra studier här vi dock ej funnit stöd för denna model; genom att blockera bildning av malonyl-CoA i b-celler kan glukos- och fett-metabolism dissocieras från varandra. Trots detta kan glukos utan något hinder stimulera insulinfrisättning, vilket betyder att en ökning av acyl-CoA ej är en förutsättning för adekvat glukos-stimulerad insulinfrisättning.
Nyligen föreslog Claes Wollheim och hans kollegor att glutamat deriverat från citronsyra-cykeln skulle kunna utgöra den eftersökta metabola signalen. Detta grundas ytterst på att mitokondriell metabolism synes vara kritisk för insulinfrisättning och att cellpermeabla former av glutamat potentierar glukos-stimulerad insulin-frisättning. Å andra sidan, om man bryter ned glutamat i b-cellen hämmas glukos-stimulerad insulin-frisättning. Mekanismen har föreslagits vara att aminosyran, som bildas i ß-cellen när denna stimuleras av glukos, transporteras ut ur mitokondrien och in i insulingranula, vilket startar exocytosen. Ett problem är att dessa resultat ännu ej har kunnat reproduceras i andra laboratorier. Exempelvis har vi själva ej kunnat påvisa någon signifikant stegring av glutamat i våra klonala b-celler efter exponering för glukos. Man har också undersökt om proteinkinas A och C kan spela någon roll vid den KATP-oberoende vägen för glukos-stimulerad insulinfrisättning. Härvidlag har inte någon entydig bild tonat fram; resultaten varierar mellan såväl olika laboratorier som olika species. Arakidonsyra, kväveoxid eller fosfatidylinositolfosfat-3-kinas tycks ej heller vara ansvariga. Jean-Claude Henquin har föreslagit att en förhöjd ATP:ADP kvot i anslutning till insulinfrisättning skulle kunna influera andra processer än att stänga KATP-kanalen och således verka på andra nivåer i insulinfrisättningsprocessen; exempelvis skulle granula kunna modifieras av en sådan mekanism det har länge varit känt att ATP krävs för exocytos.
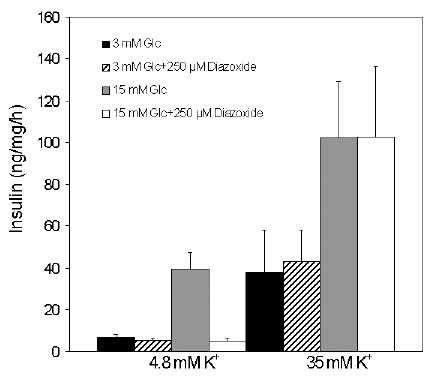
Fig. 3 Klonala ß-celler deriverade från INS-1 celler stimulerades med 3 eller 15 mM glukos i närvaro eller frånvaro av 35 mM K+ och 250 mM diazoxid. Insulin samlades under 2 h i en statisk inkubation. Diazoxid blockerar insulinfrisättning när fysiologisk K+ koncentration råder eftersom KATP-kanalen hålles öppen. I närvaro av 35 mM K+ potentierar en ökning av glukos insulinfrisättning oavsett om KATP-kanalen hålles öppen av diazoxid eller ej. Således har glukos en förmåga att frisätta insulin utan att stänga KATP-kanalen under förutsättning att Ca2+ höjes i cellen den KATP-oberoende vägen.
En titt i kristallkulan
Har de nya rönen rörande den KATP-oberoende vägen för glukos-stimulerad insulin-frisättning några implikationer för behandlingen av typ 2 diabetes? Svaret på den frågan är att i nuläget har de inte det. Emellertid är det uppenbart att upptäckten av de KATP-oberoende mekanismerna erbjuder nya måltavlor för farmakologisk behandling av den sviktande insulinfrisättningen vid typ 2 diabetes, en svikt som är en direkt orsak till att sjukdomen manifesteras. En klar fördel med att rikta in sig på KATP-oberoende mekanismer är att de är förstärkande till sin natur och tycks ej ensamma kunna utlösa insulinfrisättning. Detta står i kontrast till hur sulfonylurea verkar. Alltså, närvaro av glukos kommer att vara permissiv för en farmakologisk effekt på den KATP-oberoende vägen och risken för hypoglykemier kan därmed begränsas. Hypoglykemier uppfattas av nästan alla patienter som ett stort praktiskt problem, förenat med försämrad livskvalitet. Att identifiera metabola signaler vid den KATP-oberoende vägen för glukos-stimulerad insulin-frisättning och molekyler som modifierar denna torde vara ett högprioriterat område inom såväl grundforskning som läkemedelsindustri.
Författaren erhöll 1997 ett stipendium av Stiftelsen för vetenskapligt arbete inom diabetologi för postdoktorala studier vid Touchstone Center for Diabetes Research i Dallas, Texas. Han är nu verksam vid institutionen för cell- och molekylärbiologi, Lunds universitet. Ett särskilt tack riktas till Professor Claes Hellerström, Uppsala, och Professor Carl-David Agardh, Malmö, för stöd och uppmuntran.
För vidare läsning rekommenderas:
Newgard, C.B. and McGarry, J.D. (1995) Metabolic coupling factors in pancreatic beta-cell signal transduction. Annual Review of Biochemistry, 64, 689-719. Prentki, M. and Corkey, B.E. (1996) Are the beta-cell signaling molecules malonyl-CoA and cytosolic long-chain acyl-CoA implicated in multiple tissue defects of obesity and NIDDM? Diabetes, 45, 273-83. Wollheim, C.B. (2000) Beta-cell mitochondria in the regulation of insulin secretion: a new culprit in type II diabetes. Diabetologia, 43, 265-77. Henquin, J.C. (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes, 49, 1751-60. Mulder, H., Lu, D., Finley, J., An, J., Cohen, J., Antinozzi, P.A., McGarry, J.D. and Newgard, C.B. (2001) Overexpression of a modified human malonyl-CoA decarboxylase blocks the glucose-induced increase in malonyl-CoA level but has no impact on insulin secretion in INS-1-derived (832/13) beta-cells. J Biol Chem, 276, 6479-84. Lu, D., Mulder, H., Zhao, P., Burgess, S.C., Jensen, M.V., Kamzolova, S., Newgard, C.B. and Sherry, A.D. (2002) 13C NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS). Proc Natl Acad Sci U S A, 99, 2708-13.
En fullständig
referenslista kan erhållas från författaren.
hindrik.mulder@medkem.lu.se
