
Glukagon-sekretionen vid hypoglykemi medieras av autonoma nervsystemet hos friska individer
Av Bo Ahrén, Malmö.
Bakgrund
I den så kallade DCCT-studien visades att långt driven metabol kontroll i diabetesbehandlingen minskar uppträdandet av senkomplikationer som retinopati och nefropati men ökar samtidigt risken för akut hypoglykemi (1). Konsekvensen av detta är att hypoglykemi blir den begränsande faktorn vid intensiv behandlingen (2). För att bättre kunna förebygga risken för hypoglykemi och för att förstå faktorer som påverkar kroppens förmåga att motreglera hypoglykemin är det viktigt att förstå hypoglykemins fysiologi och patofysiologi. En sådan förståelse är också viktig för att kunna förklara varför många patienter med diabetes har en försämrad försvarsmekanism mot hypoglykemi. Tidigare studier har visat att adekvat glukagon-sekretion är nödvändig för motreglering till hypoglykemi (2-4). För att förstå mekanismen bakom försvarsreaktion vid hypoglykemi är det således viktigt att förstå mekanismen bakom den glukagonsekretion som aktiveras av hypoglykemi.
Man kan därvid tänka sig flera möjligheter. En mekanism kan vara att det sänkta blodsockret i sig stimulerar glukagonsekretionen. Detta har visats i experimentella system, exempelvis i perfunderad pankreas där låg glukoshalt i det perfunderande mediet stimulerar glukagonsekretionen (5), men om denna mekanism verkligen är av fysiologisk betydelse vid de cirkulerande glukosnivåer som förekommer vid hypoglykemi är inte fastställt.
En annan mekanism bakom den stimulerade glukagonsekretionen vid hypoglykemi kan vara en minskning av den lokala insulinnivån i de Langerhanska öarna, eftersom det låga blodsockret hämmar insulinsekretionen. Den sänkta lokala insulinkoncentrationen kan då inverka stimulerande på de närbelägna alfa-cellerna, eftersom insulin hämmar glukagonsekretionen. En sådan mekanism har experimentellt stöd genom resultat av studier där man perfunderat pankreas med antikroppar riktade mot insulin. Dessa har bundit upp det insulin som frisatts från beta-cellerna, vilket resulterat i stimulering av glukagonsekretionen (6).
En tredje förklaring bakom stimulerad glukagonsekretion vid hypoglykemi är att hypoglykemin har aktiverat det autonoma, icke viljestyrda, nervsystemet, vilket i sin tur stimulerat glukagon-sekretionen. Med det autonoma nervsystemet förstås i detta sammanhang både de parasympatiska nerverna och de sympatiska nerverna vilka innerverar de Langerhanska öarna och adrenalin frisatt från binjuremärgen. Vid hypoglykemi aktiveras såväl parasympatiska som sympatiska nerver och adrenalin frisätts från binjuren, och samtliga dessa tre delar av det autonoma nervsystemet stimulerar glukagonsekretionen (7). Om denna mekanism verkligen är av fysiologisk betydelse för glukagonsekretionen hos människa har dock inte fastställts tidigare. I djurförsök på möss har vi dock kunnat visat att aktivering av det autonoma nervsystemet är av vital betydelse för normal glukagonsekretion vid hypoglykemi genom att glukagon-sekretionen hämmas om det autonoma nervsystemet blockeras vid hypoglykemi (8). Huruvida så är fallet även på människa har vi nyligen genomfört en undersökning där vi givit farmakologisk ganglieblockad, hämmande både det sympatiska och parasymptiska nervsystemet, och undersökt om detta påverka glukagonsekretionen vid insulininducerad hypoglykemi hos friska människor (9).
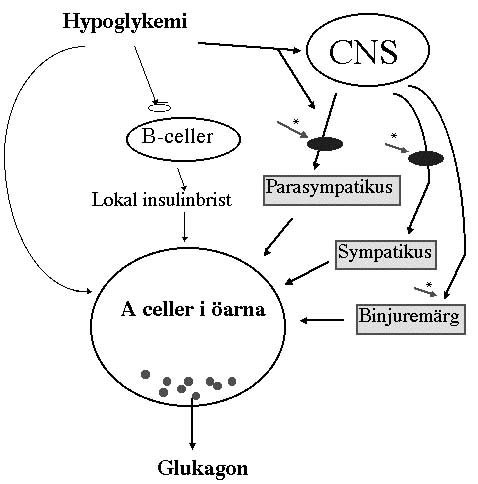
Figuren visar schematiskt tre teoretiskt möjliga mekanismer för aktivering av glukagonsekretionen från pankreasöarnas A celler (eller -celler): direkt aktivering via det låga blodsockret, indirekt aktivering via sänkt lokal insulinkoncentration i öarna i närheten av A cellerna respektive neurogen aktivering (via det autonoma nervsystemet).
Den senare kan ske via centrala nervsystemet (CNS) eller via perifera mekanismer. De tre grenar som aktiveras är parasympatiska nervgrenar och sympatiska nervgrenar respektive binjuremärgen. Dessa tre grenar av det autonoma nervsystemet aktiverar glukagonsekretionen via specifika transmittorer (exempelvis acetylkolin, noradrenalin, adrenalin och/eller neuropeptider såsom vasoaktiv intestinal polypeptid (VIP), gastrin frisättande peptiden (GRP), cholecystokinin (CCK), neuropeptid Y (NPY) eller galanin).
Det autonoma nervsystemets perifera grenar står under inflytande av autonoma ganglier, vilkas aktivitet hämmas av trimetofan. Binjuremärgen är i detta sammanhanget att betraktas som ett autonomt ganglion, som också hämmas av trimetofan. Eftersom trimetofan i denna studie markant hämmade den glukagonfrisättning som erhölls vid hypoglykemin talar resultaten för att mekanism via det autonoma nervsystemet är av avgörande betydelse.
De autonoma ganglierna är avbildade svart, och de pilarna markerade med * anger platsen för trimetofans hämmande effekt.
(ArfonadR) eller koksalt. Trimetofan gavs i ett dosintervall om 0.3-0.6 mg/kg/min genom att dropptakten för trimetofan-lösningen reglerades individuellt för varje försöksperson efter blodtrycket, som tilläts falla högst 10 mmHg. Genom att trimetofan hämmar de autonoma ganglierna erhålles en experimentsituation där exempelvis förändringar i glukagonsekretionen har medierats utan medverkan av det autonoma nervsystemet. En skillnad i glukagonsekretionen vid de båda undersökningarna som genomfördes med trimetofan respektive koksalt kan alltså tolkas vara orsakad av det autonoma nervsystemet.
Blodprov togs för regelbunden bestämning av glukagon, pankreatisk polypeptid (PP; markör för parasympatiska nervsystemet), adrenalin och noradrenalin (markörer för det sympatiska nervsystemet). För att utesluta att trimetofan i sig, oberoende av det autonoma nervsystemet, påverkar glukagonsekretionen utfördes även ett kontrollförsök där arginin (5 g) injicerades
Genomförd studie
Studien utfördes vid Klinisk Forskningsenhet Medicin vid Universitetssjukhuset MAS i Malmö och är publicerad i tidskriften Diabetes (9). Sju kvinnor med normal glukostolerans (ålder 64 år) undersöktes vid två tillfällen med en 45 min hyperinsulinemisk (serum insulin ~700 pmol/l) hypoglykemisk clamp vid vilken plasma-glukos sänktes till 2.5±0.2 mmol/l. Samtidig gavs en intravenös infusion av ganglieblockeraren trimetofan intravenöst för att stimulera glukagonsekretionen med eller utan samtidig intravenös tillförsel av trimetofan.
Studien visade att under hypoglykemin frisattes glukagon, PP, adrenalin och noradrenalin. Trimetofan hämmade hypoglykemi-inducerad ökning av glukagon med 72±17% (p<0.005). Samtidigt hämmades ökningen av PP (med 71±16%, p<0.0025), adrenalin (med 70±16%, p<0.005) och noradrenalin (med 79±21%, p<0.005).
I den andra försöksserien visades att den ökning av plasma glukagon som erhölls efter intravenös injektion av arginin inte påverkades av samtidig trimetofan-infusion (+132±30% utan trimetofan jämfört med +135±24% med trimetofan).
Diskussion
Vår studie har visat att huvuddelen (ca 70%) av hypoglykemi-inducerad glukagonsekretion kan hämmas av autonom ganglieblockad hos friska individer. Slutsatsen av studien är således att glukagonsekretionen under hypoglykemi till största delen är medierad av det autonoma nervsystemet hos friska individer. Även om dessa resultat naturligtvis måste reproduceras även på individer med diabetes leder våra resultat till frågan om försämrad funktion i det autonoma nervsystemet kan utgöra riskfaktorer för utveckling av hypoglykemi hos patienter som behandlas med insulin genom att glukagonsekretionen ej kan stimulereas i adekvat omfattning.
Man kan därvid spekulera i flera bakomliggande mekanismer, vilka kan inträda vid olika tidsperioder efter debut av diabetes. En lätt förståelig mekanism vore autonom neuropati inkluderande de autonoma nerver som innerverar de Langerhanska öarna. En sådan neuropati skulle leda till försämrad glukagonfrisättning vid hypoglykemi och därigenom en försämrad motregulatorisk förmåga. Emellertid finns försämrad glukagonfrisättning vid hypoglykemi oftast hos patienter innan klassisk autonom neuropati uppträder (10). Detta skulle tala mot en sådan koppling. Emellertid är det inte osannolikt att mer subtil neuropati än den som diagnosticeras som klinisk autonom neuropati kan finnas i tidigare skede av sjukdomen. En sådan möjlighet har nyligen visats av Bolli och medarbetare som funnit att såväl adrenalin som PP-svaret vid hypoglykemi är nedsatt hos patienter med typ 1 diabetes vilka ännu inte utvecklat kliniska tecken på autonom neuropati (11). Vidare kan en tidig, funktionell, störning i de autonoma nerverna uppträda som resultat av genomgångna episoder av hypoglykemi. Detta har kallats "hypoglykemi-associerad autonom dysfunktion" (HAAF) och beskrivits av Cryer och medarbetare (12,13).
Detta kan orsakas av att patienten genom en långt driven insulinbehandling får upprepade, kanske i sig inte kliniskt tydliga, hypoglykemiepisoder vilket leder till en funktionell hämning av de autonoma nervernas funktion och därigenom till en försämrad förmåga att senare adekvat kunna försvara sig mot hypoglykemi. Viktiga aspekter på detta tillstånd är dels att det kan orsakas av endast en episod av hypoglykemi och dels att det är reversibelt och försvinner om hypoglykemi undvikes. Fortsatta studier av detta är nu önskvärt. I nuläget kan vi sammanfatta att, även om ytterligare studier är nödvändiga för att förstå den kliniska betydelsen av våra aktuella resultat, våra resultat hittills har visat att det autonoma nervsystemet är av funktionell betydelse för den glukagonsekretion som sker vid hypoglykemi hos människa, vilket tidigare endast var känt i djurförsök.
Bo Ahrén, Doc, överläk
Institutionen för medicin, Lunds Universitet,
Universitetssjukhuset, 2050 02 MalmöReferenslista kan fås från DiabetoFax 08 34 79 55 dokument nr 93032
[Innehåll] [Redaktören] [Ordföranden] [Sett
& Hört]
[Aktuell Info]
[Redaktionen] [Arkivet] [Länkar] [Diskussionsforum] [Diabetes Update]
